1. 清空环境变量及控制台
rm(list = ls())
cat("\014")2. 加载包
如果没有请自行下载,如果基础板安装不上,可以用bioconductor安装,点我查看安装方法
library(tidyverse) #安装包
library(openxlsx)
library(pheatmap)3. 导入数据
这里需要根据自己的数据类型导入,我用的xlsx,所以导入的命令如下,当然,你们也可以用其他函数导入,只要是数据,都可以.这里,我们列是样本名,行名是基因名,不要弄错数据格式了!
dat=read.xlsx("../MergeBasisForPaper/41HubGenes_FPKM.xlsx",rowNames = T)
names(dat)
HeatmapData=dat4. 构建列(样本)分组(注释)信息
在这里,矩阵行名是样本名称,列是分组信息,不要弄错了!同理,对基因的分组也是同样的构造办法!
annotation_row1=read.xlsx("../MergeBasisForPaper/coldata.xlsx",rowNames = T)
annotation_row=annotation_row1[,-2] #根据你的数据选取,其他信息都不要
annotation_row=as.data.frame(annotation_row) #因为只有一列就是数组,所以需要强制转化一下数据类型
rownames(annotation_row) <- rownames(annotation_row1)
colnames(annotation_row) <- "Sample"
annotation_col=read.xlsx("../MergeBasisForPaper/coldata.xlsx",rowNames = T,sheet = 2) #保存与否自己决定
save(annotation_col,annotation_row,file = "../code/heatmap_annotation.Rdata") #保存与否自己决定上面的路径是我自己的路径,你别无脑复制。基础操作不会的点我学习
5. 画图与保存
期刊所需要高清图片,参数res调整,里面的参数用法不会的可以“?函数名”即可知道,如“?pheatmap”
tiff(filename = "../MergeBasisForPaper/Pictures/filename.tiff",
width = 4,
height = 2,
res = 300, # 这是DPI,期刊一般要求300
units = "in") # 这是图片长宽为英寸,还可以设置为“cm”
par(oma=c(3,3,3,3))
par(mar=c(6,6,4,3) + 0.1)
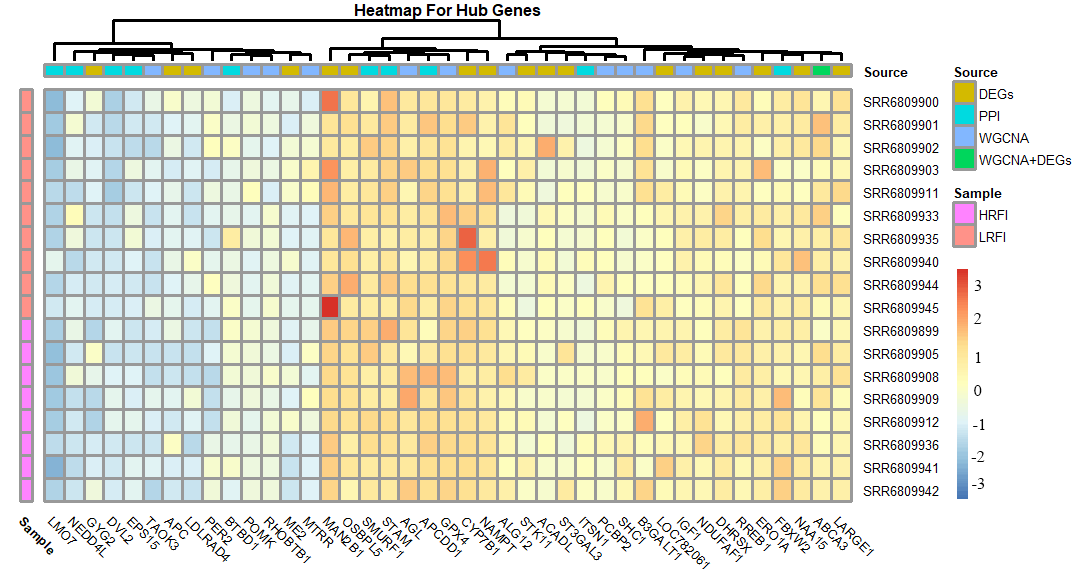
pheatmap(t(HeatmapData),
scale = 'row', # 标准化行或者列取决于你的数据格式,比如列是sample1-sample5,行名是gene1-gene5,那么标准化就“row”,也就是说我们要看同一个基因在不同样本间的表达量,这样归一化做出来的图才有意义
annotation_col = annotation_col, # 注释文件
annotation_row = annotation_row, # 注释文件
cluster_rows = F, #聚类与否
cluster_cols = T, #聚类与否
#cellwidth = 10, #每个小格子的宽度
#cellheight = 10, #每个小格子的长度
angle_col = 315, # 横轴标签旋转角度
#clustering_method = 'average',# 聚类方法
#cutree_rows = 2, # 切分聚类数量
#cutree_cols = 5, # 切分聚类数量
display_numbers = F, # 是否显示数字
fontsize_row = 3, # 行数字大小
fontsize_col = 3, # 列数字大小
fontsize = 3, # 数字大小
treeheight_col = 5, # 聚类树高度
main = "Heatmap For Hub Genes", # 起个名字
legend_breaks=NA, # 图例
color = c("green","red"), #自定义颜色,但是这个自定义出来的效果可能没有默认的好
treeheight_row = 5, # 聚类树高度
annotation_colors = c("red","blue","black","yellow")# 注释的颜色
)
dev.off() # 关闭画图设备6. 结束
好了,这就是热图的做法,不懂的可以留言讨论
最后,贴一张效果图