1. 概述
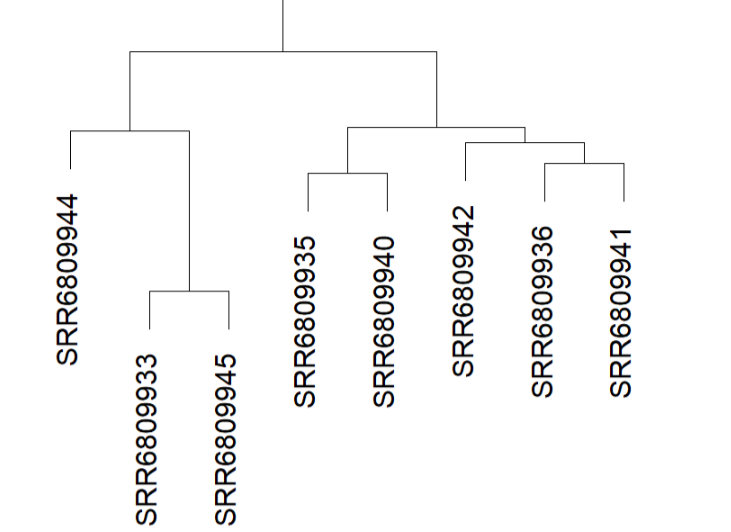
上一期的文章基于R语言的样本聚类-1用R语言实现了基于聚类距离的聚类树的实现,最终的结果是得到基于某个表型(比如基因表达趋势变化)的分组,在同一个分支(一类)上的两个样本有更多的相似之处,上一期的最终效果如下图:
而用于检测或发现样本间相似性的方法还有主成分分析法,相关性分析方法等。基于样本件的相关性,也可以将样本进行聚类。主成分分析的原理推荐一篇知乎文章前往学习。今天,我们就利用R语言实现这一个分析过程并且可视化。
2. R语言实现
2.1 加载包及导入数据
本次所用到的包ggcorrplot+openxlsx,不会安装包的戳我学习,里面有安装包的办法,这里就不多说了。
rm(list = ls()) # 清空环境变量
library(ggcorrplot) # 加载包
library(openxlsx) # 加载包2.1.1导入数据
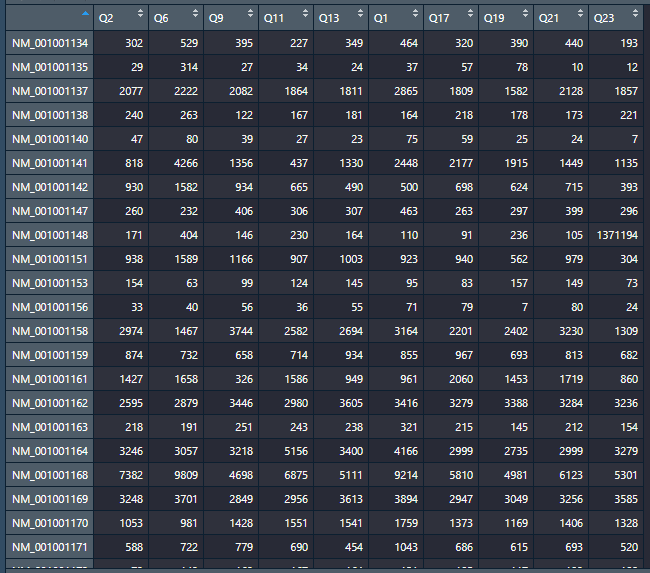
load('MydataforStringTie.Rdata') # 该数据是我的一个数据集,里面包含很多数据,你不必纠结他是干什么的含有什么的,你只需要知道你需要一个下面的“dat"内容的数据框即可!
dat=GeneCounts # 赋值
head(dat,6) # 查看数据框前6行,其实默认的也是6行
names(dat) # 查看数据框的名字数据格式如下:
2.1.2 计算相关系数及其显著性
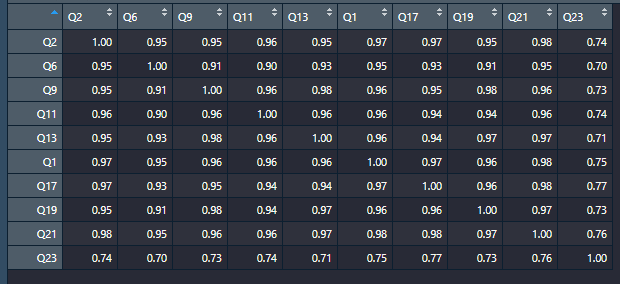
corr=round(cor(dat[,]),2) # Calculate the correlation coefficient
p.mat=cor_pmat(dat)# Calculate the significance of correlation coefficient样本相关性结果如下:
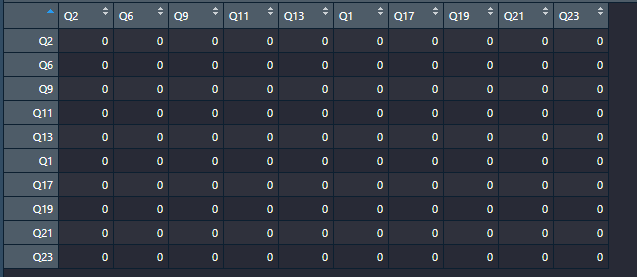
由于数据量的问题,我的数据没能检验相关额显著性,如下:
2.2 结果可视化
准备好数据后,利用ggcorrplot函数可以对相关性的分析结果进行可视化,代码如下:
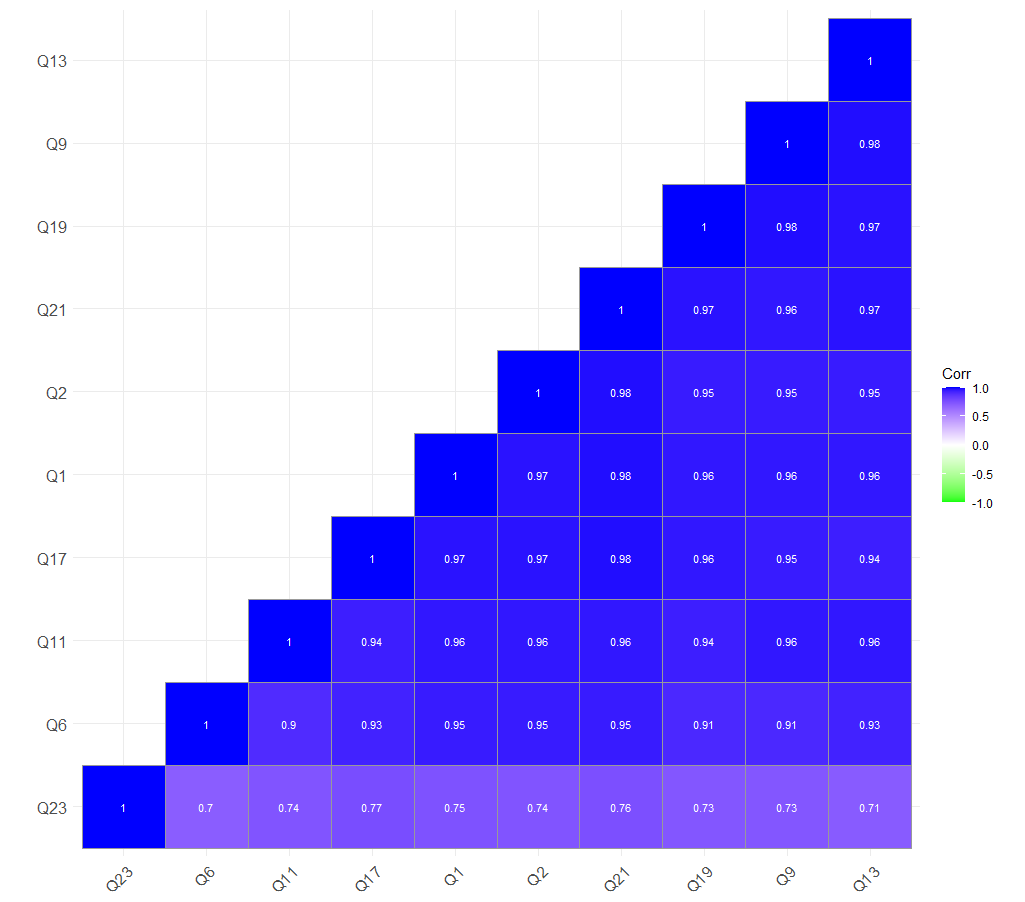
ggcorrplot(corr,
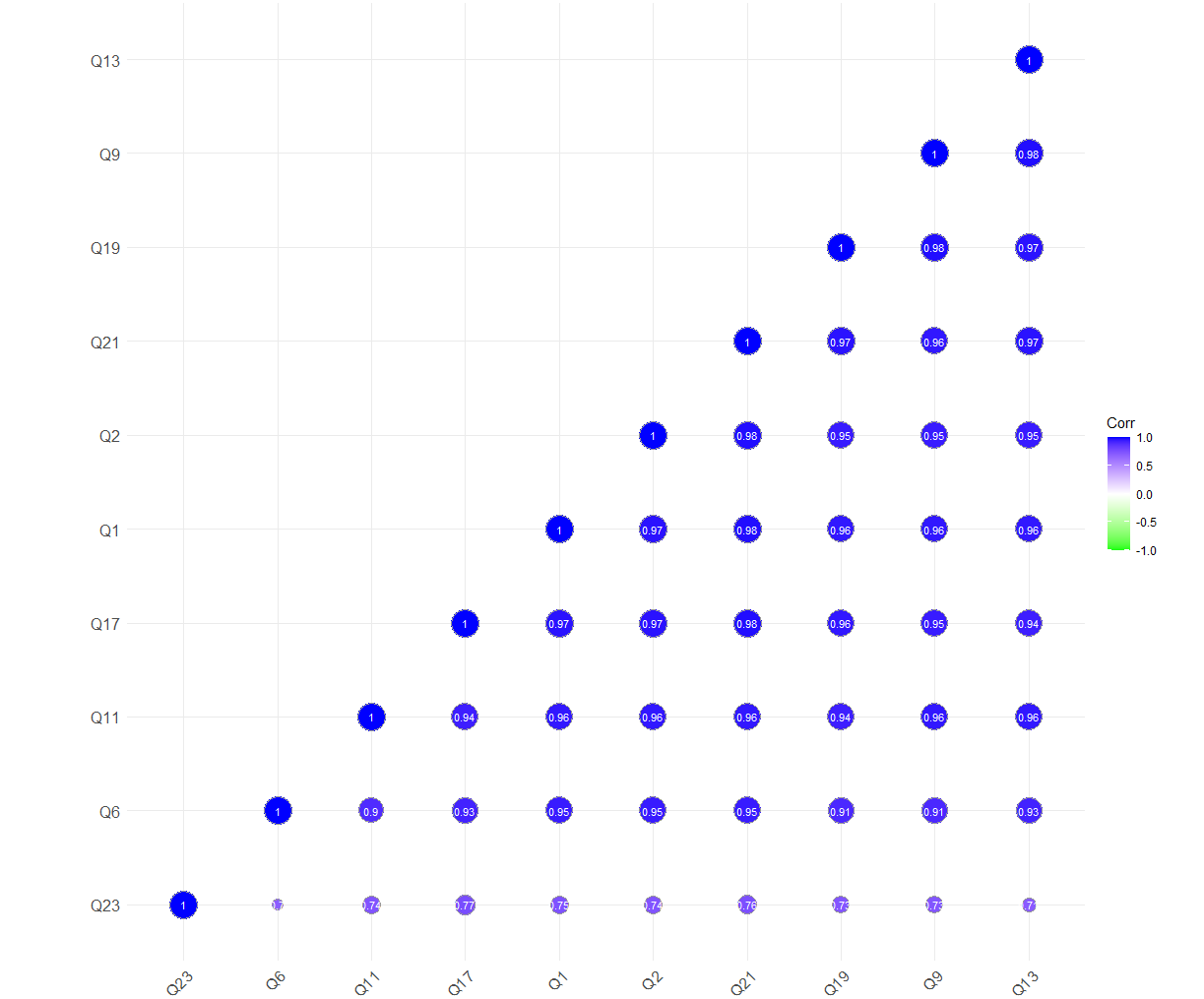
method = 'square',# 显示样式,“circle"是第二张图的样子
hc.order = TRUE, # 是否聚类
type = 'lower', # 上下三角
outline.color = 'gray60', # 框颜色
colors = c('green','white','blue'), # 图颜色
lab = TRUE, # 是否显示数字
lab_col = 'white', # 数字颜色
lab_size = 3, # 数字大小
p.mat = p.mat, # 填充
insig = 'blank',# 显著的黑色填充
show.diag = TRUE, # 只显示对角线一侧
digits = 2 #小数保留位数
)可视化结果如下,可以看得出来我的样本Q23是有些问题的,如果需要考虑离群值,这个样本可能是候选之一:
由于这个分析基本都是在数据预处理的时候用,花里胡哨的那些我就懒得上了,如果喜欢花里胡哨的可以去看这篇博文。
好了,今天就更新到这儿,下一期我们更新基于PCA分析的样本聚类。
记得打赏哟,白嫖我来你家吃饭!



