1. 简要概述
1.1 WGCNA用来做什么
Weighted Gene Co-Expression Network Analysis(WGCNA, 加权基因共表达网络分析),主要用于鉴定表达模式相似的基因集合(module)。解析基因集合与样品表型之间的联系,绘制基因集合中基因之间的调控网络并鉴定关键调控基因。
1.2 基本过程
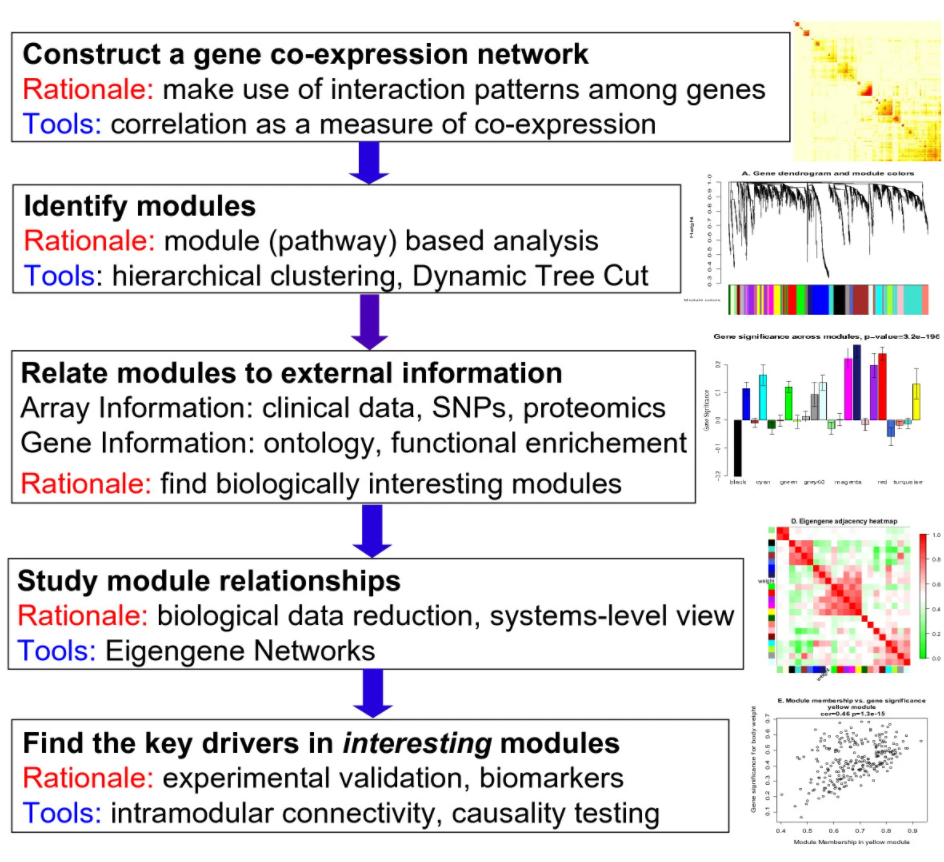
详细过程请点击,这是原图链接,追求高清的可以去看看。总结下来,应包含以下几个过程:
A、构建基因关系网络
B、构建基因模块
C、筛选关键模块
D、鉴定关键基因
2. 数据准备
在WGCNA分析的过程中,我们所用到的文件有两个,其一是基因表达文件,其二是表型文件。
这这里,基因表达文件我们只能用表达量,不能使用计数矩阵,表达量比如RPKM/FPKM/TPM等均可,也可以用DEseq2标准化后的数据。
同时,表型文件是我们研究的表型分组数据,比如数量性状[又叫连续型变量]的产肉量、产奶量、跳高等。或分类变量[又叫离散型变量],这一类数据比如月龄、产犊胎次、蛋壳颜色、药物效果等。
2.1 基因表达文件的准备
今天这个部分我用好基友**书哲**大神的数据来进行演示。
2.1.1 安装R语言及Rstudio
详细的安装方法我就不介绍了,这里给大家提个醒,也是经常出现坑的地方!
第一:他们两安装的路径不能含有中文路径,出现中文路径估计某天你就会发现它不工作了!!!切记!
第二:先安装R内核,我一般选择清华镜像,点我安装;安装完成后你再安装Rstudio,选择免费版就好,一般科研工作者足够用了!点我安装
本次WGCNA分享我所用的内核+Rstudio版本如下:
2.1.2 包的安装
在安装R包时,会出现各种版本不兼容的问题,为此Bioconductor完美解决了这个问题。首先,我们先安装这个万能的管理包:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install() # 里面可以指定对应的版本,如 “version = "3.13"”然后就可以愉快的安装包了,下面贴上我在使用的包,你们可以酌情安装,在以后使用的时候直接使用以下命令安装即可:
BiocManager::install("Packagename")我所用包:
BiocManager::install(c("org.Bt.eg.db","DESeq2","dplyr","RColorBrewer",
"genefilter","GO.db","topGO","GSEABase",
"clusterProfiler","fgsea","tidyverse","ggpubr",
"pheatmap","DESeq2","gplots","GOplot","ggplot2",
"enrichplot","UpSetR","VennDiagram","openxlsx",
"WGCNA","gmodels","ggcorrplot","scatterplot3d"
))加载某一个包用下面一个命令:
library(Packagename)2.1.3 数据导入
Ok,在上面的准备工作做完了以后,我们开始导入数据。在开始一个项目之前,我个人习惯性进行如下操作:
rm(list = ls()) # 清空环境变量,这个的作用不用我多说吧
getwd(); # 获取当前工作路劲
workingDir = "." # 设置当前工作路径为“.”,目的是方便我们要陪你过Tab键补齐和切换路径
setwd(workingDir)加载包
library(openxlsx)
library(WGCNA)个人习惯性导入数据前都会用如下代码先指定一下,防止将数据中的字符串当做因子处理
options(stringsAsFactors = FALSE)# 不要将字符串当做因子处理导入数据
dat=read.xlsx("../Filename.xlsx") # 根据自己的喜好选择合适的导入方式,如TXT,tsv,csv格式等
dim(dat) # 查看dat的维度
head(dat) # 查看dat的前6行导入后如下:
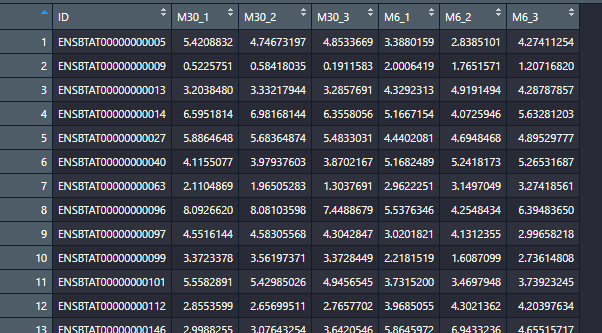
这个实验室1因子2水平3个重复的设计,不懂的自己去翻生物统计的书。
2.1.4 ID转换
通常,我们常见的基因ID是gene symbol,即类似CCK /CDH2/P53/IGF1/FN1之类。上述的基因ID看起来是Ensembl 注释文件中的命名风格。这里我们转换一下ID。
加载包
library(org.Bt.eg.db) #这是物种牛上的注释包,其他物种的需要下载对应注释包
library(GO.db)
library(topGO)
library(GSEABase)
library(clusterProfiler)
library(fgsea)
library(tidyr)提取需要转化的ID并转化为字符串
trans_id <- dat$ID
symbol=as.character(trans_id)转换
ids_to_symbol <- bitr(symbol,
fromType="ENSEMBLTRANS", # 从ENSEMBLTRANS转化至下面的两种ID
toType=c('ENTREZID',"SYMBOL"),
OrgDb="org.Bt.eg.db")转换后结果如下:
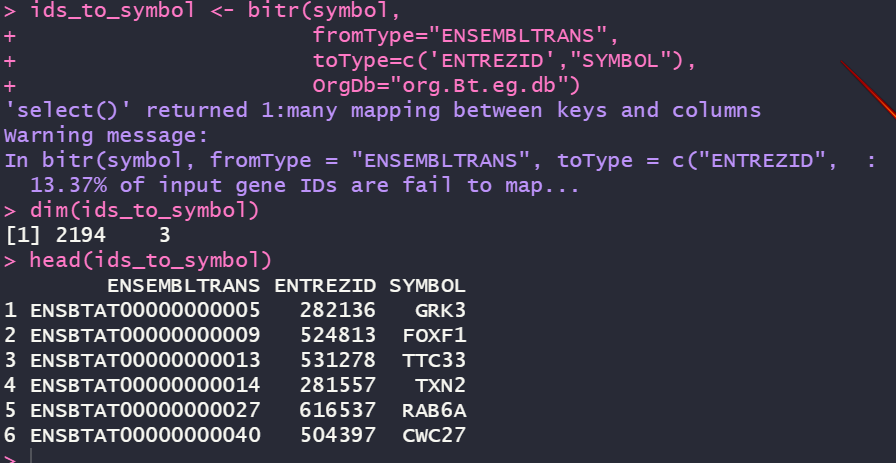
提示有13.37%的基因名转换不成功!这个数据如果偏大可能是你的注释包可能弄错了,我这边这么高的原因可能是黄牛属与水牛属的区别。当然,这一步可以去David网站上进行转化。
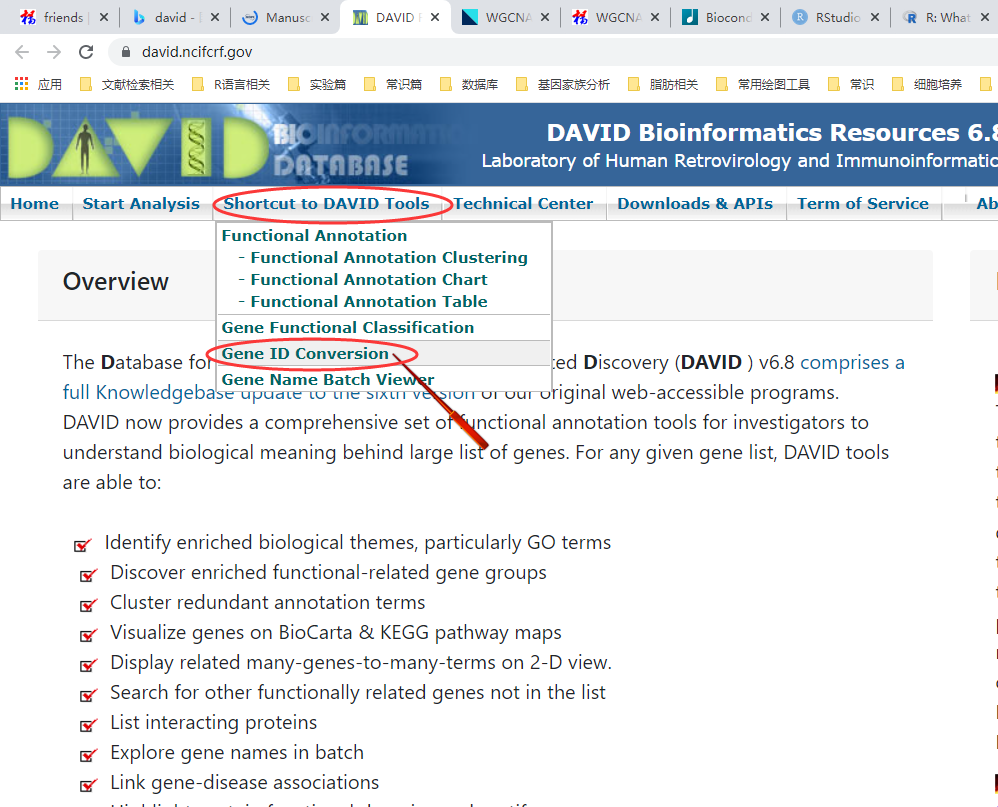
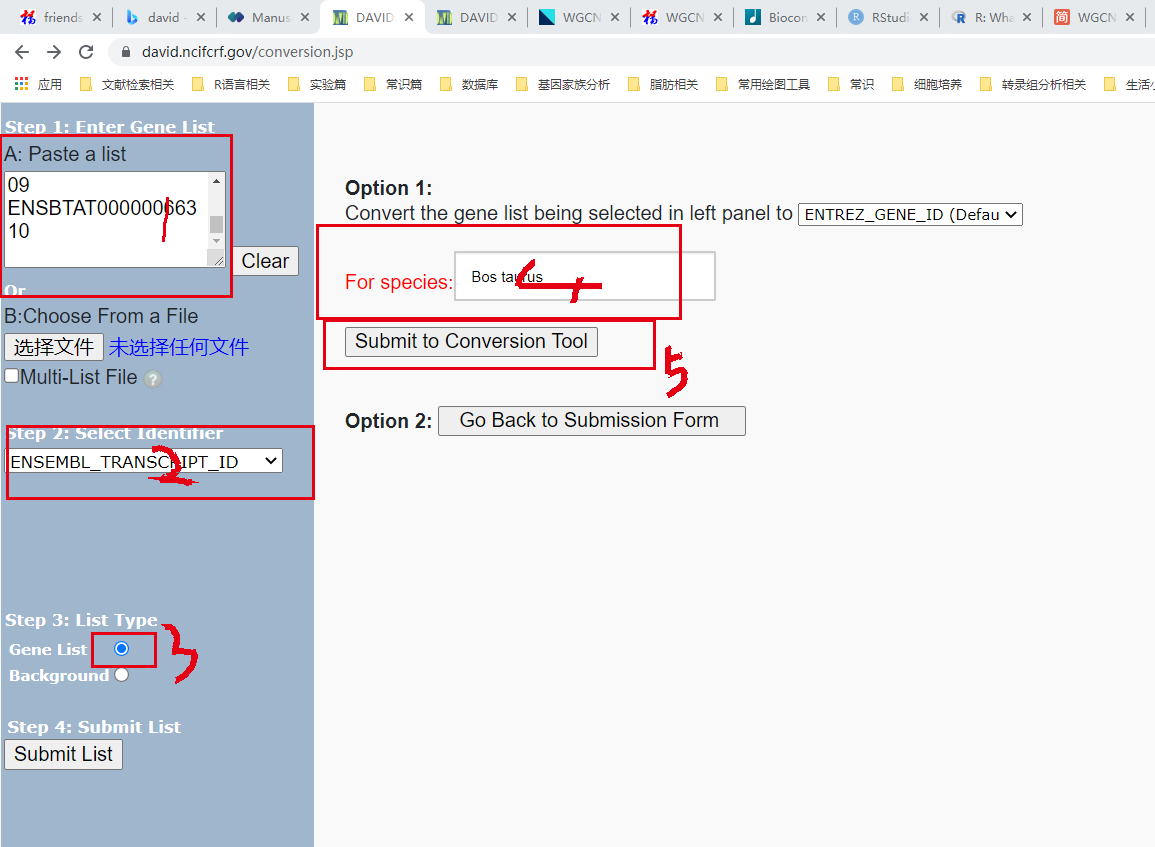
折腾一番后,结果如下:
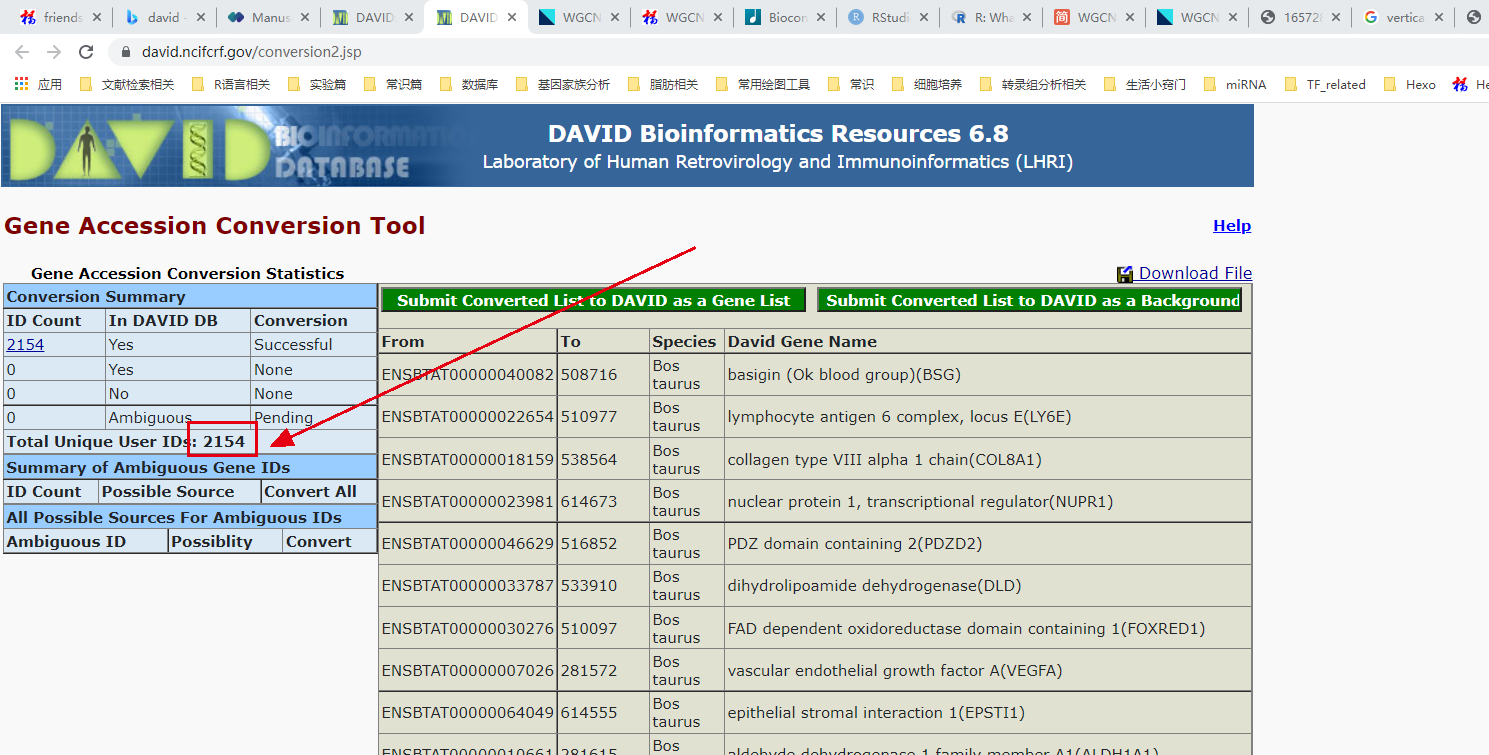
两种方法都是2100多个,选其一即可。
2.1.5 合并转换结果
直接上代码:
dat=merge(dat,ids_to_symbol,by.x="ID",by.y="ENSEMBLTRANS") # 合并两个数据框
head(dat)
rownames(dat) <- dat$SYMBOL #将SYMBOL命为行名合并结果如下图,但是当我们命名的时候出现了以下的的问题:行名重复!

仔细检查了下,原来数据是基于转录本的定量,所以有重复很正常!那么我们就用Ensymbol ID作为行名,这个转化的结果保存起来后面备用!当进行了行名指定后,还是出现了这个行名重复的问题,如下:
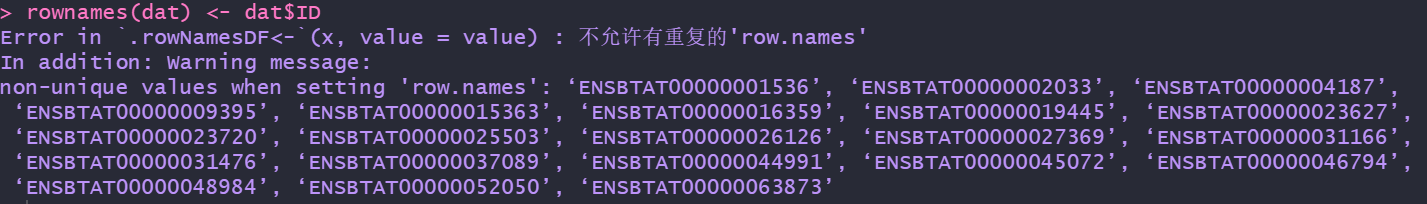
写到这里,我突然想到了我之前做比对和定量的时候,用的是UCSC的参考基因组+注释文件,那里面的转录本都有不同的编号,不会出现这个问题!既然这样,这2100多个基因都是差异转录本,那么我继续去掉重复,然后再命名。
在去掉重复之前,我们先看看有多少个重复,代码如下:
y=as.data.frame(table(dat$ID))结果如图:可以看出其实重复的并不是很多,这里有两种办法,一种是手动去重,也就是留下差异加大、表达量高一些的基因,这种需要一个个去检查。这里我为了保证数据更可靠,就直接检查去除。
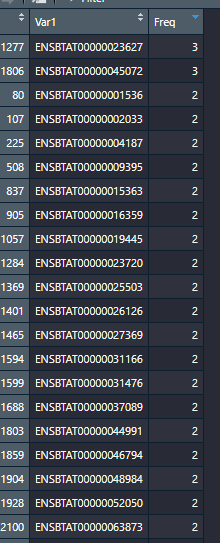
去除的话直接用数据框的操作方式就行了,但当我准备这样干的时候,问题来了,看图:同一个Ensymbol的ID,又对应了不同的Symbol ID [我崩溃了!!!]。其实刚刚我同样用了symbol ID做了一次,发现大小写字母的也算是重复,哥哥我也是崩溃的!比如BoLA,BOLA也算重复!
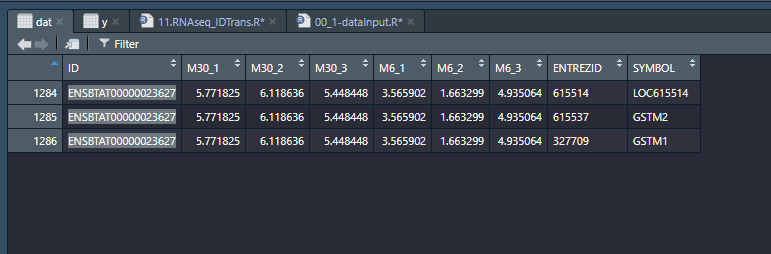
那好,既然这么刚,那我们直接去Excel把它搞一下!先保存!
write.xlsx(dat,"../Expdat_real.xlsx")保存后的如下:
打开操作它:先按照symbol升序,随后进行条件格式高亮重复值,再进行颜色排序,效果如下:然后修改就是了
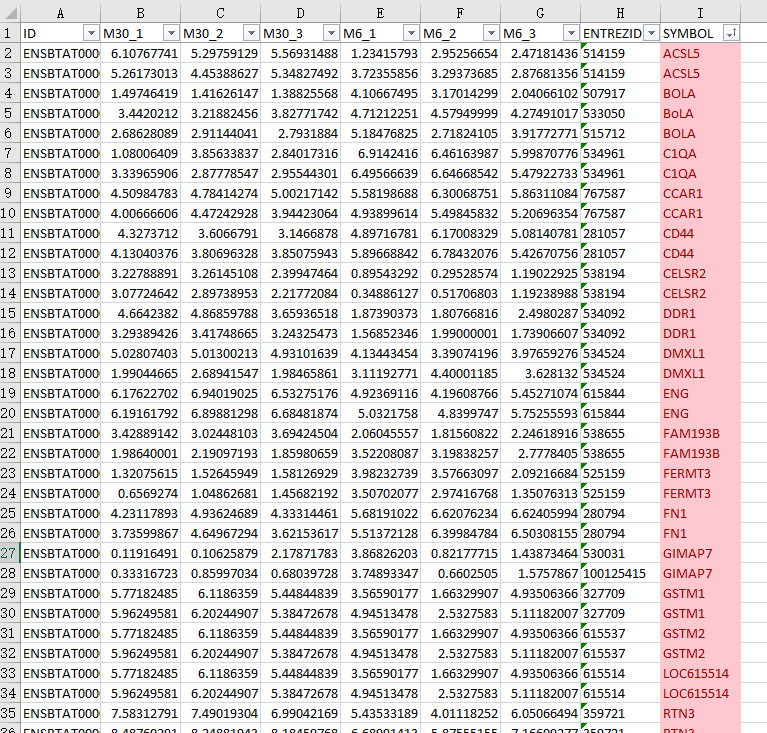
然后重复的全被我添加了后缀,如图:
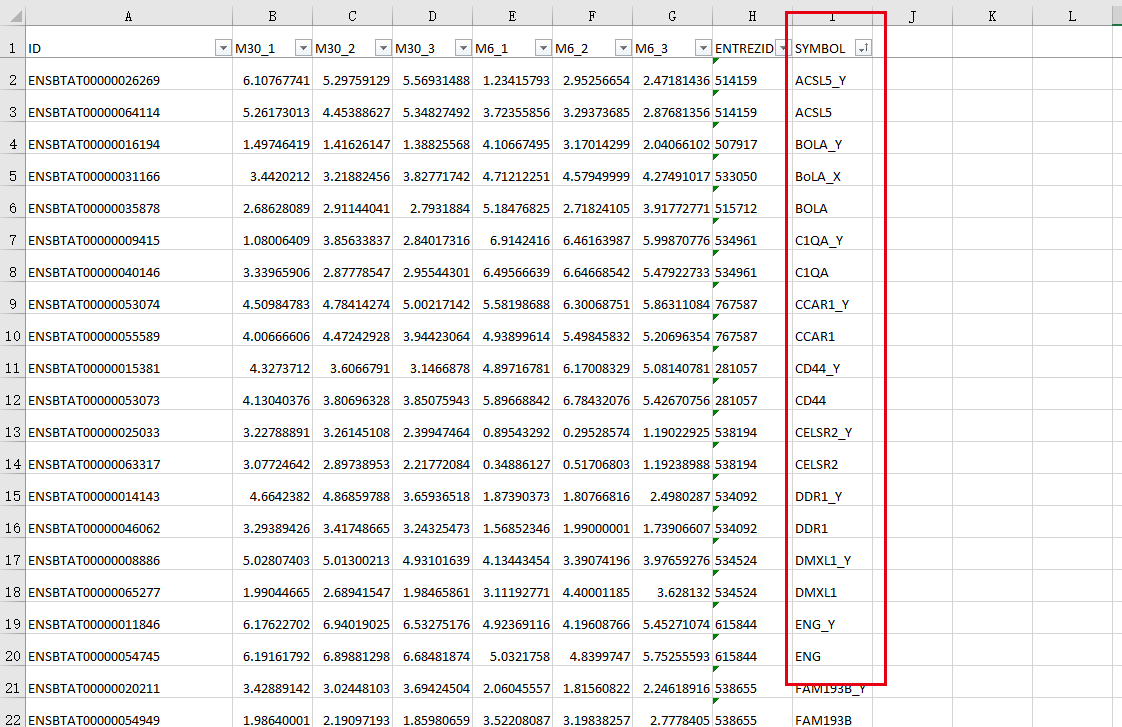
2.1.6 表达量适合度检测
经过上面的处理,我们终于拿到了想要的数据,重新导入,并留下我们想要的数据
dat=read.xlsx("../Expdat_real.xlsx")
head(dat)
rownames(dat) <- dat$SYMBOL #给行命名
colnames(dat) #查看列名
all_exp = dat[,2:7] # 最后保存只有表达量数据的数据框这一步得到的数据格式如下:
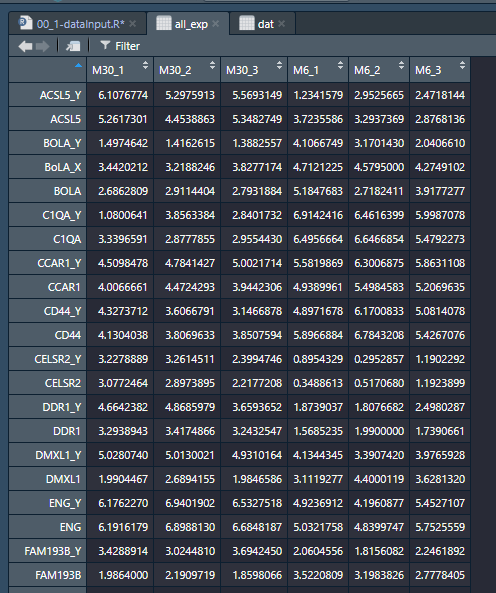
上述我们拿到了一个表达矩阵,包含了约2千个观测值,往往在我们的研究中,可能这里得到的是数以万计的基因,比如我自己弄的就有3万个左右,那么这么多的基因,我们肯定没法全部考虑进去,需要进行一定的处理后保留上千个基因,比如3千,5千,1万啥的,这里就需要有相应的处理方式保留想要用于WGCNA分析的基因。
基于上述的需求,我们这里引入一个统计量-绝对中位差(Median Absolute Deviation,MAD)。

绝对中位差是一种统计离差的测量。而且,MAD是一种鲁棒统计量,比标准差更能适应数据集中的异常值。对于标准差,使用的是数据到均值的距离平方,所以大的偏差权重更大,异常值对结果也会产生重要影响。对于MAD,少量的异常值不会影响最终的结果。
由于MAD是一个比样本方差或者标准差更鲁棒的度量,它对于不存在均值或者方差的分布效果更好,比如柯西分布。说到这里,我们之前常常用**平均值±1.5倍标准差**来剔除离群值,但是这样是存在缺陷的,可能直接抹除了我们想要的某些效应。
所以我们引入R语言中MAD函数来剔除离群值。话不多说,看代码:
m.mad <- apply(all_exp,1,mad)# 对矩阵每一行求MAD,apply函数不会用的去查查
y=as.data.frame(m.mad) # 将上述得到的vector转变为数据框
dataExprVar <- all_exp[which(m.mad > max(quantile(m.mad, probs=seq(0, 1, 0.25))[2],1)),]#不知道你们看到这个会不会懵逼,如果懵逼,先去看看quantile函数,然后再去看看seq函数,再去看看如何取一列数中的第几个,再看看如何选取数据框中如何筛选满足慢些条件的位置。里面第二个“1”是可以变化的,一般情况下,我们取0即可。在这里,我们取“0”最终结果如下:
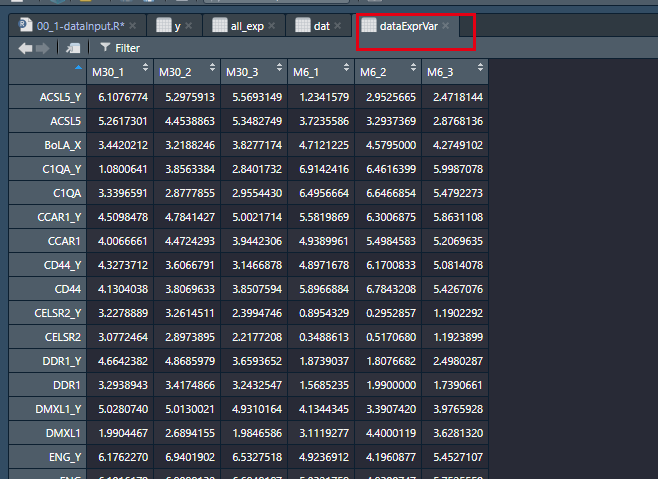
经过上述操作,我们得到一些表达量相对较高的1645个基因。
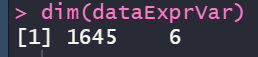
2.1.7 NA值检测
先将数据框转置再变为数据框
datExpr0 = as.data.frame(t(dataExprVar[,]));检测NA值及低于样本阈值的样本
gsg = goodSamplesGenes(datExpr0, verbose = 3); # 检测是否有基因和样本有较多的缺失值
gsg$allOK # true为OK
table(gsg$allOK)
# 如果最后一条语句返回TRUE,所有基因都通过了检测。如果不是,我们就从数据中删除违规的基因和样本。
# 若存在较差的样本或者基因,经过下面的代码剔除
if (!gsg$allOK)
{
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")));
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
table(gsg$allOK)2.1.8 聚类法检测样本离群值
接下来我们对样本进行聚类(与后面的基因聚类相反),看看是否有明显的离群值。
直接上代码:
sampleTree = hclust(dist(datExpr0), method = "average"); # 计算样本之间的聚类距离
tiff(filename = "../result/1.clusterTree.tiff",
width = 8,height = 6,units = "in",
pointsize = 5,res = 600) # 设置画图函数,一般按照期刊要求走即可,我们要求稍微高一点
par(cex = 2); # 对文字大小进行设置
par(mar = c(4,4,4,4)) # 页边距
plot(sampleTree,
main = "Sample clustering to detect outliers",
sub="",
xlab="",
cex.lab = 1.5,
cex.axis = 1.5,
cex.main = 1.5)
dev.off()图片可以看得出来我们的数据还是很好的,组内组间区分得很明显。这样我们就不用去掉异常的样本了。

如果有离群的样本,可以用下面代码设置相应参数Cut它
h=300; # 比如我想要出掉M6_2样本,我只需要h=40即可!
cutHeight=h
abline(h = h, col = "red");
clust = cutreeStatic(sampleTree, cutHeight = h, minSize = 1) # 剩下的个数
table(clust)
#以下代码是去除离群后重新组合其他样本
keepSamples = (clust==1) # 保留哪些
datExpr = datExpr0[keepSamples, ] # 保留keepSamples这些
nGenes = ncol(datExpr) # 列出基因数量
nSamples = nrow(datExpr)# 列出样本数量2.1.9 构建表型数据
一般情况下,如果表型数据较少的,我会选择直接用代码构建:如下
traitData = data.frame(sample=colnames(dat[,2:19]),
RFI=c(rep(0,5),rep(1,5),rep(0,5),rep(1,3)),
ADG=rnorm(18,1.1,0.3));上述代码看不懂的可以略过,可以直接用下面的方法:
直接Excel构建好,然后导入,Excel格式如下:
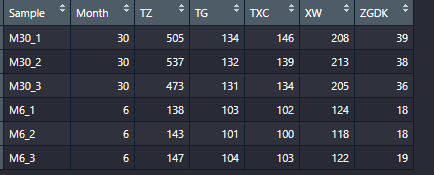
然后无脑上代码,如下:
traitData = read.xlsx("../Trait.xlsx")
dim(traitData)
names(traitData)
Samples = rownames(datExpr) # 提取行名(即样本名)并赋值
traitRows = match(Samples, traitData$Sample)# 匹配切割后的样本,返回一组数组(能够匹配上的样本位置)
datTraits = traitData[traitRows, -1]; # 构建能够匹配的数据框,去掉第一列的样本名
rownames(datTraits) = traitData[traitRows, 1]; # 给表型数据添加行名(样本名)
collectGarbage()# 回收内存如下就是构建好的表型数据
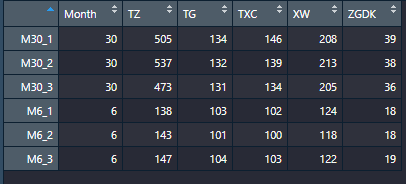
然后便是重新画聚类图和性状的热图了(表型与样本树状图的关系可视化),代码如下:
sampleTree2 = hclust(dist(datExpr), method = "average")
traitColors = numbers2colors(datTraits, signed = FALSE);
tiff(filename = "../result/1.clusterTree with RFI relationship.tiff",
width = 4,
height = 2,
units = "in",
pointsize = 5,
res = 300)
plotDendroAndColors(sampleTree2,
traitColors,
groupLabels = names(datTraits),
main = "Sample dendrogram and trait heatmap")
dev.off()效果图如下:
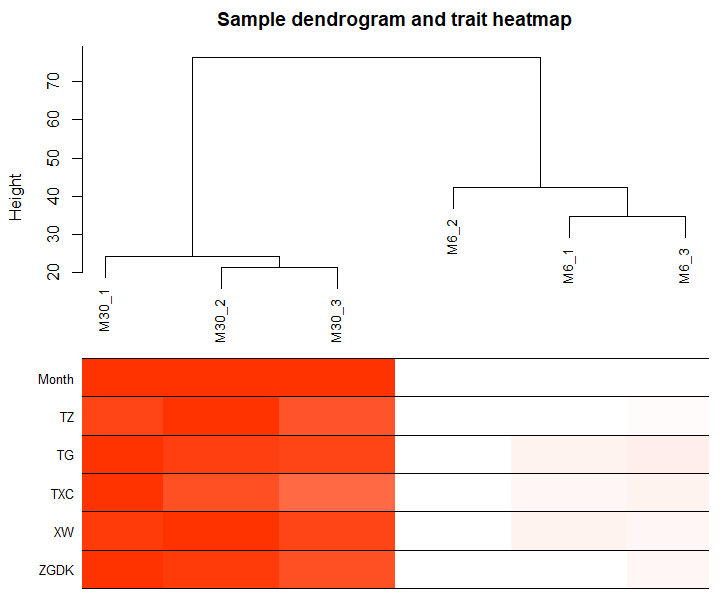
在这里,颜色深浅代表性状平均值大小,其中红色是高平均值,白色是较低平均值,灰色是缺失值。
我简单说下热图怎么制作的吧,比如我们的表型数据”TZ”,是按照上面的表型数据框里面的列(同一个表型在不同样本中的观测值)进行归一化,归一化后在进行排序比较。热图的制作可以去看看我的另一篇博文用R语言如何画一张漂亮的热图。好了,这里就扯到这儿啦!
2.20 保存表达数据及表型数据
在我们构建完成后,我们需要保存表达矩阵+表型数据用于后续的分析。
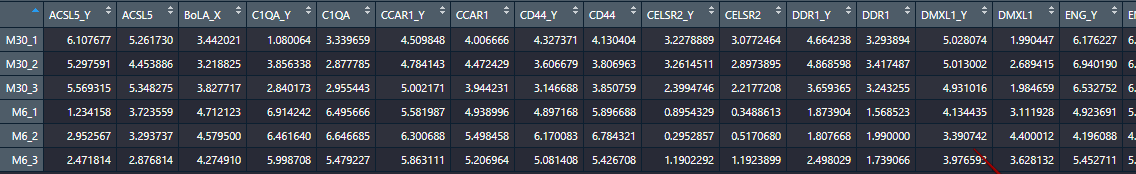
表达矩阵如下:
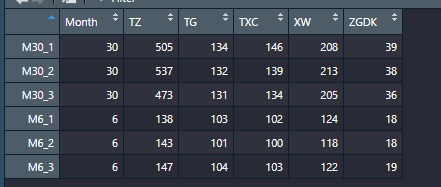
表型数据如下:
保存代码如下:
save(datExpr, datTraits, file = "../result/1.Deodunum-01-dataInput.RData")



